







NOVEL ALGORITHMS AND SOFTWARE FOR
BIOLOGICAL SEQUENCE ANALYSIS
by
William Casey Bullock
A thesis
submitted in partial fulfillment
of the requirements for the degree of
Master of Science in Computer Science
Boise State University
May 2013
c 2013
William Casey Bullock
ALL RIGHTS RESERVED
BOISE STATE UNIVERSITY GRADUATE COLLEGE
DEFENSE COMMITTEE AND FINAL READING APPROVALS
of the thesis submitted by
William Casey Bullock
Thesis Title: Novel Algorithms and Software for Biological Sequence Analysis Date of Final Oral Examination: 13 December 2012
The following individuals read and discussed the thesis submitted by student William Casey Bullock, and they evaluated his presentation and response to questions during the final oral examination. They found that the student passed the final oral examination.
Timothy Andersen, Ph.D. Chair, Supervisory Committee
Amit Jain, Ph.D. Member, Supervisory Committee
Owen McDougal, Ph.D. Member, Supervisory Committee
The final reading approval of the thesis was granted by Timothy Andersen, Ph.D., Chair, Supervisory Committee. The thesis was approved for the Graduate College by John R. Pelton, Ph.D., Dean of the Graduate College.
dedicated to Alicia, Raiden, and Lyncoln
ACKNOWLEDGMENTS
The author wishes to express gratitude to the Defense Threat Reduction Agency. This work has been partially supported by contract number W81XWH-07-1-000, DNA Safeguard. The author would also like to thank Dr. Timothy Andersen for his guidance and assistance.
ABSTRACT
Bioinformatics is a broad realm of research in which Computer Science has much to offer. Collecting, sorting, and analyzing statistical information for DNA and protein sequences is difficult due to the sheer amount of available data. Tools have been created to do this, but they have generally been limited by speed or robustness. In addition to analyzing the statistical properties of biological sequences, it is also important to model and understand their chemical and physical properties. A number of valuable software tools are available for modeling and predicting the properties of biological sequences in Computational Chemistry, including molecular docking, and homology modeling. These tools are typically command-line driven, have a steep learning curve, and generally must be used in conjunction with other programs to extract the desired information. The average chemist or biologist is not well-versed in Computer Science principles nor command-line tools and involved scripting, and therefore finds it difficult to realize the full potential of tools that are available to them.
The work completed here assists the field of Bioinformatics with two software packages for biological sequence data analysis. One is CseqStat, which processes and gathers statistical data from large repositories of genome sequence data. The algorithms I have developed in CseqStat help speed up processing large amounts of sequencing data for nucleotides and proteins in the NCBI database. This application is robust in its abilities, finding the frequency of all sequences of a given length, in a reasonable time frame, determined by the length and the amount of input data, storing the results in an efficient manner, and providing a mechanism for quick and easy retrieval of such data.
The second utility is DockoMatic, a multi-faceted software package offering the ability to run high-throughput experiments, structure creation, and molecular docking. I have collaborated with members of the Department of Chemistry and Biochem istry at Boise State University to gain valuable insight to improve existing methods.
DockoMatic was developed to allow a user to invoke and manage large numbers of molecular binding calculations, linear and cyclic peptide an a log structure creation, and molecular modeling experiments on a single computer or cluster. Specifically, Docko Matic was created to
• automate peptide-based lig and creation based on single-letter residue codes,
• automate Auto Dock job creation, submission, and management for high-throughput docking experiments,
• automate competitive binding experiments,
• track multiple jobs in real-time on a cluster,
• organize results in a useful manner,
• provide an intuitive GUI,
• automate cyclic peptide analog creation, and
• provide a simple interface for creating molecular models.
TABLE OF CONTENTS
ABSTRACT………………………………………..
LIST OF TABLES……………………………………
LIST OF FIGURES…………………………………..
1 Introduction………………………………………
2 CseqStat…………………………………………
2.0.1 Motivation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.0.2 Design. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.0.3 Performance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.0.4 Historical Data-Results. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3 DockoMatic ………………………………………
3.0.5 Motivation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.0.6 Design. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.0.7 Performance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.0.8 Publications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.0.9 Research Projects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4 Conclusions ………………………………………
REFERENCES………………………………………
LIST OF TABLES
2.1 CseqStat processing and reprocessing times for NCBI data . . . . . . . . . .
2.2 Historical data from CseqStat processing of NCBI databases. . . . . . . . .
LIST OF FIGURES
2.1 Search results for sequence a a cg. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.2 Choose new data base from directory tree. . . . . . . . . . . . . . . . . . . . . . . .
2.3 List of available databases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.4 10 results returned . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.5 10 additional results returned. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.6 Returned results not batched. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.1 Docko Maticinter face . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.2 Lig and CCMWF docked to recept or Ac-AChBP . . . . . . . . . . . . . . . . . .
3.3 Competitive binding experiment results. . . . . . . . . . . . . . . . . . . . . . . . .
3.4 Lig and screated with Docko Matic . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.5 TIM’s first window. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.6 TIM’s second window. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.7 TIM’s third window. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
3.8 TIM’s fourth window, looking at the. pap alignment file . . . . . . . . . . . .
3.9 TIM’s fourth window, looking at the. pir alignment file. . . . . . . . . . . . .
3.10TIM’s fifth window;the model calculations are done . . . . . . . . . . . . . . .
CHAPTER 1
INTRODUCTION
Absent and rare sequences have a number of potential uses including barcodes for labeling biological materials, such as DNA reference samples, since they either do not occur in nature, or occur very infrequently. Genetic samples are used by many agencies, such as the military and law enforcement for varying reasons, such as identification of suspects and victims in criminal cases and paternity testing. Although collecting and storing DNA is quite common, and the methods have become better, there are no significant new technologies to protect the many samples from errors in the laboratory; the most common being cross-contamination. The CseqStat project was initiated to identify absent or prime sequences not seen in nature. Their absence in nature makes them ideal candidates for the tagging of DNA samples. Studying DNA lead us to also look at protein sequences. The goal of
CseqStat for the DNA Safeguard project was to monitor biological databases in order to identify absent and rare sequences, as well as how these change over time.
Prime DNA and protein sequences are interesting to study in order to answer the question of why they do not occur, and how they may interact with other molecular structures under various differing environmental conditions. In fact, it has been conjectured that one reason they do not occur is due to their toxicity. Recently certain prime sequences, or nullomers, have been identified that could lead to new cancer therapies [1]. Preliminary tests show that these nullomers have lethal effects on cancer cells, while their effects on normal cells decreases over time. The processing power gained from computer clusters and cloud computing has
helped computational modeling to become increasingly useful in analyzing physical and chemical properties. Ligand to receptor binding interactions can be modeled with software programs such as AutoDock. AutoDock is powerful and popular, although to setup and run jobs, then collect the results, it requires a considerable amount of user time [2].
It is also interesting to study how known ligands may interact differently with receptors when specific amino acids are changed. Molecular modeling is a field of computational chemistry that allows the creation of three-dimensional structure models for macromolecules based on the molecular template of a known molecule.
DockoMatic was developed to simplify job submission and management of virtual high-throughput molecular docking and modeling experiments. This has the potential for broad impact in areas of drug discovery and molecular modeling. DockoMatic automates high-throughput screening of ligand to receptor binding interactions via a convenient Graphical User Interface (GUI), facilitating creation and management of AutoDock jobs.
DockoMatic includes the ability to automatically create peptide ligand protein database (.pdb) files and invoke and manage AutoDock jobs on a cluster of nodes or a single computer, with functionality to evaluate secondary ligand interactions [3]. Also, included in DockoMatic’s arsenal is the ability to create cyclic peptide analog structures, and a wizard to guide a user through the steps of creating homology models for peptides or proteins. Results are easily evaluated, as DockoMatic collects and summarizes them automatically.
CHAPTER 2
CSEQSTAT
2.0.1 Motivation
The main repository of biological data is The National Center for Biotechnology Information (NCBI); the single largest repository for nucleotide and protein data [4]. The total monthly set of DNA and protein data to be processed and analyzed from the NCBI database is close to 300 GB zipped, which amounts to approximately 500
GB of raw sequence data, with more being added daily. To put this in perspective, the complete works of Shakespeare uses only 5 MB, so this is approximately 100,000 times more data.
Daily updates make it desirable to have a timeline of the changes, since NCBI does not provide archived snapshots nor a mechanism for easy access to historical data.
In order to generate the statistics, I have helped develop CseqStat, a sequence processing tool, which allows one to find the number of occurrences of sequences for a given length of DNA or peptide. Currently, due to memory restrictions, data for sequences up to length 17 are able to be efficiently generated, however, the software does allow for processing up to length 32. The specific objectives were to
• Store monthly backups of the NCBI data.
• Generate statistics for each backup.
• Make processed data available to others.
2.0.2 Design
CseqStat is a suite of tools to evaluate and compress large amounts of data, and make the results easily retrievable and manageable. It consists of a processor, reprocessor, and utilities to create and access numerous custom file types in order to provide efficient storage. In order to condense such data, I designed two file types to store processed results; the most efficient depends upon the number of prime sequences found for a given length. The NZ, or non-zero file type only stores the frequency of sequences that occur, thereby saving space by not storing the sequences that do not occur. This is beneficial when there are large numbers of prime sequences, and makes generating and searching for those sequences fast. Since the strings for a given sequence take 8 bits per character, each sequence string is converted to a 64-bit numeric key, which only takes two bits per character of storage when processing DNA.
This realizes a storage savings by a factor of four. Using bits also enables the reverse complements of the DNA sequences to be easily produced with simple bit manipulations. In order to accurately obtain the frequency, each sequence is read in the forward direction, and then again in the reverse direction; the reverse direction being composed of the complements of the original nucleotides. Bit shifting and complementing are much faster than traversing a character array, or performing time consuming calculations with multiplication each time a new character in a sequence is read.
Working with proteins does not require reverse complements to be calculated, although it does pose its own set of intricacies using the bit representation. Since there are only 4 nucleotides for DNA, they are easily stored with 2 bits. On the other hand, there are 20 amino acids for proteins, which is not a power of 2. In order to represent the 20 amino acids, 5 bits are required. While this does cause some space to be wasted, it is still better than using a string representation when storing results, since, as stated before, 1 character consumes 8 bits of storage. This convention is also scalable to any number of input characters. For instance, if using the entire alphabet as the base set, as would be the case to study documents, such as articles, for frequency of certain words, 5 bits per character is still used. In fact, 5 bits would cover a base set up to 32 characters. By utilizing binary representations of string sequences, processing of nucleotide and protein data is sped up by at least an order of magnitude. By designing efficient file formats, the amount of storage needed for the statistical data generated is significantly reduced.
File Formats
There are two binary file storage formats utilized with CseqStat: Non-zero (NZ) and All-counts (AC). For longer length sequences, it is necessary to split up processing across multiple nodes due to memory constraints. Both file formats begin with a certain amount of meta data including length of sequence that was processed, base sequence, number of splits used for processing, and which split it is. For instance, if processing length 16, the job would need to split up four times, with each split being written to a separate binary file. With length 16 split 4 times, four binary files are used, each approximately 8 GB in size.
NZ Format
The NZ file format utilizes a binary array with each bit signifying if a given sequence was found or not. The binary array takes a total of 4N bits where N is the length of sequence being processed. This allows quick retrieval of prime sequences as it is only necessary to shift bits to see if given sequences have been found or not. This
file format saves space by recording only the count for each sequence that was found, rather than having to store each count consecutively for each possible sequence of a given length, which could result in many counts of 0 unnecessarily taking up space on the disk. The binary array is used to calculate the sequence value. For instance,
if processing DNA data for length 4, a binary array of length 4 would be used, which would contain a total of 256 bits, or 4 long long values. With length 4, there are a total of 44 possible sequences. In order to find all primes of length 4, it is only necessary to read in the binary array from disk. Then shift and compare each bit, calculating and printing the values when a 0 is encountered. If printing the counts for the found sequences, the same process is used, with the exception of reading in the next count when a 1 occurs. In this case, a file pointer is held at the next count value in the file.
The NZ file format is also useful for condensing results when storing DNA data, taking advantage of the fact that each sequence has a reverse complement. It is then only necessary to store a single count for each sequence and its reverse complement, which reduces the total binary file size by almost half. Not exactly half, since some sequences are palindromes, and the full binary array still has to be stored.
AC Format
The AC file format is useful when there are larger numbers of found sequences. If all sequences are found, the count for every sequence is already stored, so there is no reduction in space by using the NZ format, it actually ends up taking more space. Although the storage needed for the binary array is minimal in comparison to
the amount of space needed for the counts of the sequences themselves. One other benefit to the AC format is the ease at which it can retrieve the results; there are fewer calculations necessary to do so, since only a numeric value needs to be incremented to obtain the key, rather than calculating it outright. However, this only applies to DNA data, since the proteins still require more calculation to determine each consecutive key, due to the gaps in the numerical representation of such keys.
General Data Handling Conventions
The software is built upon a virtual base DB Object class that defines the foundation to process and reprocess data. Each inherited object has the ability to generically process Fasta or Genbank files, and store the data as NZ or AC files. Fasta and Genbank files are comprised of a series of sequences. Each sequence is preceded by header meta-data containing information about the sequence and a marker to identify the start. The two files mainly differ with respect to the meta-data they hold; Genbank files use a different set of tags to identify more detailed properties of the sequences. CseqStat is built to be generic, able to process the two files using the same methods for both. A utilities class provides the ability to convert character sequences to and from their binary numeric key equivalents, along with the rest of the general data handling at the sequence level, such as computing bit masks and generating reverse complements.
Originally, the software used a hash map to hold the keys and counts in memory, but testing proved it to be extremely slow compared to a standard array. This convention stores only the number of occurrences of each key, in which the array indices serve as the key values. This makes incrementing counts very efficient, as a single operation is used, rather than a lookup into a hash table, in which more overhead is involved.
Processing
Sequences are processed in a manner that yields all subsequences of a given length. For instance, processing sequence “atgcta,” into length 4, results in atgc, tgct, and gcta. Including the reverse complements of these sequences yields the additional sequences of tagc, agca, and gcat.
Processing begins with a pointer to a Sequence object, which is read in from a Fasta or Genbank file. Once the sequence is obtained, each character is converted to a binary representation, base 4 for DNA. Upon subsequent characters, the binary key is shifted the appropriate number of bits left, 2 for DNA, and the new binary character is added to the key with an OR operation. Once the appropriate number of characters initially populate the key, a count for the first key is stored in the sequence array. The following keys are stored upon each subsequent character found, since the length has already been populated, and previous characters are just shifted off the key. Since the key is composed of 64 bits, a simple bit mask is used, for the length desired, to obtain the actual key. When the processor begins, a table is created that contains the binary representation of each character in the base sequence to provide a single lookup operation to convert characters to their binary counterparts. The Fasta and Genbank data sometimes has characters that are neither nucleotide nor proteins.
When such a character is encountered, the sequence starts over upon the next valid character. Printing options include the ability to print only primes, only found sequences, all sequences, or to print nothing at all. Printing nothing at all is useful if the resulting binary files are to be used for reprocessing, or to be used for gathering different types of results later. It is also possible to not save binary data, which would speed up the job somewhat, as the data does not need to be written to disk. This scenario is only useful if one of the printing options is enabled.
When processing for sequences of longer lengths, it is sometimes necessary to split up the jobs to avoid using all available memory, which would cause disk swapping, thereby causing the processing job to perform extremely slow. Any number of splits can be used, however it is generally better to use powers of 2, which helps distribute the sequences more evenly. Splitting jobs can also be useful with regard to speed, and can realize a linear speedup if being used on a cluster of computing nodes. This is the preferred and intended use of the software, as it is designed for use with multiple nodes for quick processing of large amounts of data.
During testing, it was found that the read and write operations to and from disk were the bottleneck for the projected speed increases. After thorough testing, it was determined that there was an optimal read/write buffer size that was different for a parallel virtual file system as opposed to a standard EXT file system used on Linux [5]. The optimal buffer size was found to be 1 MB, while the standard buffer size was only 4 KB. Great improvements in overall running time were realized by modifying these buffer sizes within the code.
Reprocessing
In order to facilitate reprocessing existing results for shorter length sequences, it is necessary to separately keep track of each sequence that occurs after an invalid character, and the first sequence after the meta-data. This “first key” provides continuity, otherwise the resulting data would be inaccurate. For instance, processing sequence “atgcta” for length 4, results in atgc, tgct, and gcta, where the “first key” is atgc. To reprocess these results into length 3, using the naive algorithm of scanning each of the keys from head to tail, yields atg, tgc, tgc, gct, gct, and cta. Notice the duplicated sequences tgc and gct.
However, if the original sequence is processed for length 3, the result is atg, tgc, gct, and cta. The naive method causes an extra tgc and gct to be counted. To avoid this, only the last N characters of each key are used, where N is the reprocessing length. In this example, using the last 3 characters of each key yields tgc, gct, cta. Now the results lack one sequence. This is where the “first key” comes into play. For the “first key,” the entire sequence has to be examined, similar to the naive method. In the example, the “first key” was atgc. By using all characters of the “first key,” and the tails of the rest, we now have the correct result: atg, tgc, gct, and cta.
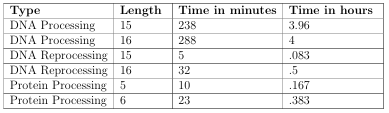
Reprocessing is useful when there exists binary files resulting from initial pro cessing jobs. These binary files can be reprocessed for various differing scenarios. One is to use results from processing jobs of large sequence lengths to obtain results for smaller sequence lengths. This can dramatically speed up the time necessary to obtain results. For instance, the results for sequence length 15 can be used to obtain results for all lengths less than 15 without the necessity of reading all information in the original Fasta files, and the reprocessing times get shorter with smaller length sequences. Processing length 15 for all Fasta data takes approximately 4 hours, while
reprocessing length 15 to length 15, to account for reverse complemented sequences, takes approximately 5 minutes. However, there is one caveat, if the amount of data for a given sequence length does not fit into memory, the results have to be written to disk in buckets, one per split, to later be gathered at the end of the reprocessing job.
This is currently necessary for lengths larger than 15 on the Boise State Genesis cluster. For instance, length 16 uses approximately 32 GB of memory, which exceeds the available RAM on each node. So, the results must be stored to disk first, then read in at the final step of reprocessing. This sounds worse than it is, since the original processing job takes almost the same amount of time as 15, due to splitting the jobs accordingly, while the reprocessing time takes approximately 35 minutes, which is still dramatically less than original processing.
To efficiently reprocess large amounts of data, it would be infeasible to attempt to store the data to be reprocessed, as well as the reprocessed data, in memory at the same time. This is avoided by using a virtual reprocessing mode that only stores in memory the pertinent information of the data to be reprocessed, while the actual sequence and count data is read from the disk as needed. This allows only the reprocessed results to be stored in memory, making it possible to handle large amounts of data.
There are numerous printing options available to reprocessing, which mirror the print options when processing. Perhaps more useful than the printing options when processing is the option to only print the results from a given binary file. This makes it possible to walk through the results of a processing or reprocessing job to print the results without the added overhead of storing sequences in memory and performing the other functions involved in processing and reprocessing. An Iterator class was created to improve reprocessing, which generically allows the use of different binary file types. This allows the code to be simplified in the respect that it does not have to perform different actions based on the input file type.
Web Interface
A client-server application was also created to allow access to processed data. This tool provides a distributable client GUI that connects to a server, making processed data available to the masses. It is written in Java to be platform independent and makes use of Java’s Remote Method Invocation (RMI) technology, which allows the client to make method calls that reside on the server, resulting in a client with a small footprint. The server interfaces with the data using the Java Native Interface (JNI), leveraging the data structures and some code from the reprocessing and processing pieces that were written in C++.
The client GUI provides access to the data in two main ways: search database for a specific sequence in which its frequency is displayed, and search database for the sequences that occur within a specified frequency range.
If searching a database for a specific sequence, the grid display will show one line with the results as in Figure 2.1. If the sequence is not found, the software returns quickly with a frequency of 0 if using the NZ file type for the database, as NZ file types strength is providing fast results for primes.

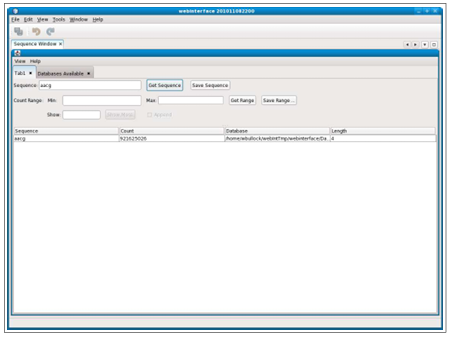
Multiple tabs can be used to show results from multiple searches. Separate tabs can also be used to display search results from different databases. Switching databases is easy to do by using the “View” drop-down menu, which gives the options:
• Switch database
• Create a new tab
• Show available databases
Figure 2.2 shows an example of the directory tree used for selecting new databases.

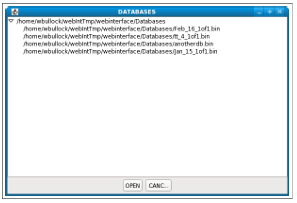
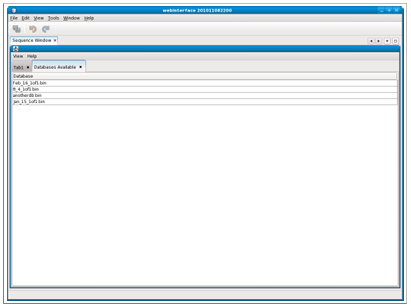
When viewing the databases currently on the filesystem by selecting the “Show available databases,” a new tab is automatically opened and the databases are shown in list format (Figure 2.3).

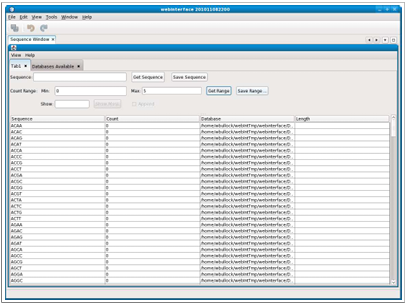
Searching for ranges of counts sometimes produces more results than can easily be displayed. The interface offers the ability to limit the number of results returned. When this option is used, results are returned in batches. A text box titled “Show” is available in which the number of results returned in each batch can be specified. The default is 50 results if the text box is left empty. Figure 2.4 shows an example of the results returned when the batch number is 10.

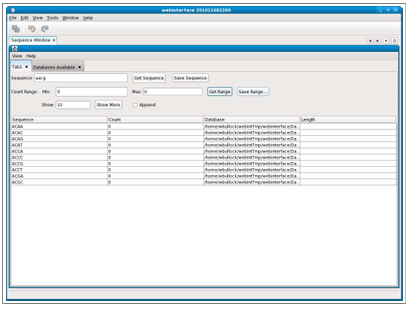
Then, the selection of the “Show more” button will display 10 additional results, fetched from the server. There are two display options when using the batch mode. If the “Append” checkbox is selected, each batch is appended to the list of results, as shown below in Figure 2.5, and if not selected, each batch replaces the previous set of results.

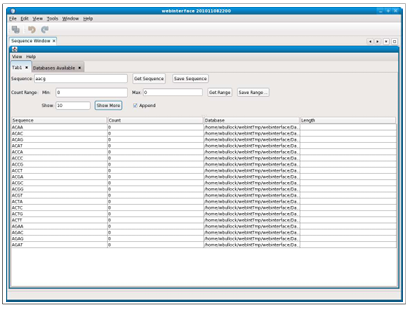
Figure 2.6 shows the results of a search when not using the batch mode and there are too many sequences to display. Note the scroll bar on the right side of the display window, which can be used to view all the results even though they do not fit on the current screen.

2.0.3 Performance
Before CseqStat was operational, processing all of the NCBI data took approximately 32 hours for length 15. Due to the various enhancements, such as bit manipulations, and efficient use of data structures, this time has been significantly reduced, especially when reprocessing is being used. Table 2.1 outlines the various speeds for different types of jobs used on the entire set of Fasta data from NCBI; DNA processing, reprocessing those results, and protein processing.

2.0.4 Historical Data- Results
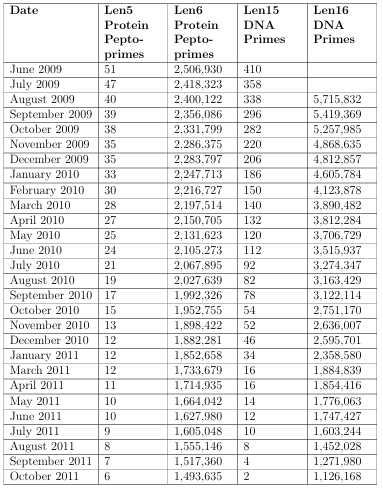
Table 2.2 shows the results from processing the NCBI databases beginning in June of 2009, and ending in October of 2011. Information is added to the databases daily, resulting in fewer primes as time goes on. Notice the trend; as more data is added, fewer and fewer primes exist. More information about the specific primes can be found at http://biotech.boisestate.edu/bioinformatics/nullomers.private.

CHAPTER 3
DOCKOMATIC
3.0.5 Motivation
DockoMatic was developed to aid the study of physical and chemical properties of statistically interesting biological sequences. For example, peptides derived from the venom of snails of the genus Conus have demonstrated potential as potent ligands for many biological receptors [6,7]. It has been challenging to understand how these peptides function by traditional wet bench experiments. Computational modeling has become a useful tool to study peptide ligand interactions with large biological receptors [8].
Many programs exist to simulate the atomic interactions between a ligand and a receptor [2,9], with AutoDock being one of the more widely used tools among them [2,10]. AutoDock ranks ligands by estimating binding interaction energy between the ligand and receptor [11], using the AMBER force field, as well as linear regression analysis, for its computations. For the analysis of a receptor and a single ligand, this works well. Since it is necessary to run ligands individually through AutoDock, high-throughput screening of peptide ligands binding to a protein receptor can be labor intensive. It is also time consuming to manually analyze the AutoDock output
results file to confirm ligand interactions. DockoMatic organizes results from molecular docking experiments automatically.
Secondary ligand interactions can also be evaluated, and single-letter amino acid abbreviations can be supplied in strings and used to automatically create peptide ligand .pdb files.
Another useful feature of DockoMatic is the ability to conduct experiments with analogs of protein structures. That is, to replace sections of the protein with other amino acids to see how a given ligand may dock differently. To create these cyclic peptide analogs, I have integrated Treepack to predict how mutated side-chains are
oriented in relation to the backbones they attach to [12]. DockoMatic also provides homology modeling capabilities, or the creation of a protein structure for a sequence where the structure is not known, with a user-friendly wizard TIM (Timely Integrated Modeller). This enables virtual experiments to be conducted that would otherwise be impossible.
The ability to generate homology models is a useful enhancement to ease an otherwise tedious and time consuming process. The software package MODELLER has been leveraged for its comparative modeling functionality [13]. In order to produce a model, a known structure of similar sequence is used as a template for
the new structure to be formed. This template structure is either already known, or is found via a similarity search of sequences, generally in a sequence database. The search returns sequences of known structure and similar composition to the original sequence. Next, MODELLERcreates an alignment from the original sequence and the
template using its align2d() command, creating PIR and PAP formatted files. Manual modifications can be made to the PAP and PIR alignment files after being determined by inspecting them. Once the alignment is agreeable, the sequence and alignment are used to create 3D model(s) with MODELLER’s automodel class. Typically, several models are generated and evaluated to determine the best fit. The most suitable model can be determined by the molpdf, DOPE, and GA341 scores, as well as using a 3D structure display program such as PyMOL [14].
Many protein complexes are formed with disulfide bonds present in the structure. This adds a layer of complexity in the generation of such models. DockoMatic includes the ability to specify residue positions in which disulfide bonds are used in the final model. Without DockoMatic, there is a steep learning curve in homology modeling. In
order to use MODELLER effectively, scripting knowledge has to be gained, as well as the intricacies of the software itself. For users without such skills, this process can take months. A user can become comfortable with DockoMatic’s wizard TIM in less than a day. DockoMatic also makes it possible to run all separate modeling jobs in parallel with the use of a cluster and no additional requirements of the user.
3.0.6 Design
Molecular Docking
Intuitive GUI
DockoMatic was designed to have an intuitive and relatively simple interface for chemists or biochemists with limited Computer Science training. It does not require command-line processing nor involved scripting. The interface has been designed to help guide the user through the requirements for a successful AutoDock job creation, submission, and analysis, or homology model creation. The GUI has been designed with NetBeans [15] to provide a robust interface, and allow functionality such as separate window panes for different components and the ability to manipulate these windows for customization of the desktop. The interface is comprised of 3 separate components that can be detached, resized, or closed from view entirely. Users can set up the windows according to their preferences, and to fit their workflow. DockoMatic’s default layout places the user required items in the input window pane as the main focus, using the central and upper left portions of the GUI. The job information or management grid pane is on the bottom, and the output messages pane is to the right.
The main input window pane is where all information for a docking job is entered. It begins with the output directory. The user clicks on a button and navigates to the directory where the results will be deposited. If no output directory is specified, it defaults to the current working directory for the user when the GUI is started. The user can either select a single .pdb file, or input a string of amino acids in the ligand box. For high-throughput screening, instead of entering an individual ligand, the user may enter a file name and check the box for “Use Ligand List File.” The file name must refer to an input file that contains either a list of amino acid strings, or paths to existing ligand .pdb files. The user selects both the receptor and box coordinate files in a similar manner to the ligand. Users may also specify a secondary ligand or a file containing a list of secondary ligands to model how an additional ligand may bond in the presence of the first ligand.
After entering these items, the user can press the “New Job” button to initialize Auto Dock jobs. This populates the management grid with a list of all jobs. The number of jobs created is equal to the cross product of the ligands, receptors, and box coordinate files. The job management window contains a list of all jobs, and provides control via a pop-up window with four options, or direct manipulation of the text fields. The management grid lists the output directory path, job number, ligand specified or path to .pdb file, path to receptor, path to box coordinate file, secondary ligand or path to secondary ligand file, and the current status of the job. Once jobs are started, the status of each job in the window is automatically checked every ten seconds. Each job to be manipulated can be highlighted by the user and the pop-up options are available with a right-click of the mouse. The options include Start, Delete, Analyze, and to view results with PyMOL (requires PyMOL installation) [14].
To ameliorate job management, the ability to group jobs in different tabs within the Job Management window is available. By default, all jobs are sent to the tab that is currently being viewed. A check box is beneath the “New Job” button that populates the job list in a separate tab. Multiple CPU cores per node can be utilized, even while running on a single workstation by using the “Jobs per Node” text field. The default is one.
All jobs are sent to swarm to be queued to the cluster, however, if using a single machine, you must make sure the custom swarm file provided with DockoMatic can be found in the PATH variable. The user also has the option to create their own swarm script to be used for job submission. The messages window pane displays information regarding job status and miscellaneous output, providing state information for jobs. Figure 3.1 displays an image of the DockoMatic GUI after being used to create a homology model. Note the path of the resulting model shown in the “Ligand” text field. The same model would result if the receptor homology model option was used, the difference being the text field in which the path to the model is shown.

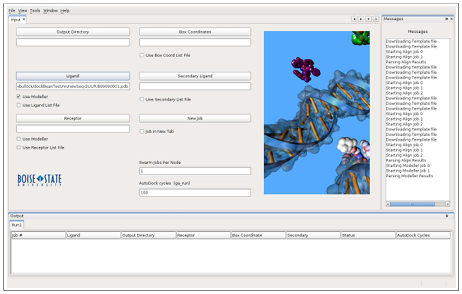
AutoDock Job Creation and Management
While DockoMatic significantly reduces the time required by the user to create and submit jobs to AutoDock, there are three things the user must provide. These include:
1. the ligand .pdb file or sequence
2. a receptor .pdb file
3. a user defined template .gpf file
The template .gpf input file only needs to define the grid box coordinates for the region of interest. DockoMatic automates creation of the fully prepared .gpf file, which includes: 1) The specific atom types to be calculated as maps by AutoGrid, taken from the already prepared ligand .pdbqt file, 2) The location of the prepared receptor .pdbqt file, and 3) Specific grid box coordinates. Automating the creation of the fully prepared .gpf saves the user time, as this is typically done manually using AutoDock Tools. User specified Cartesian map coordinates presented in the template .gpf file are required to determine the area of interest on the receptor. However, DockoMatic hides the preparation of the ligand and receptor, as well as the creation of the prepared .gpf file, from the user. Receptor and ligand .pdb files are converted into the .pdbqt file format during the submission phase. The “docking parameter file” must also be created and a box coordinate file must be prepared. These files are automatically prepared via MGLTools [16] when using DockoMatic.
AutoDock was not specifically designed to deal with the rigors of high-throughput binding experiments by itself, as it does not directly accommodate combinations of different ligands, receptors, and grid box locations. It is also not straightforward to setup jobs for simultaneous binding of multiple ligands to a receptor. DockoMatic overcomes this limitation by using lists of ligands, receptors, and box coordinate files. Jobs are automatically created for each possible combination of inputs. For example, a list of five peptide ligands, one receptor, and two box coordinate files generates (5 X 1 X2) =20 separate jobs, all of which can be run concurrently if enough nodes or processors are available. The GUI provides the ability to manipulate the individual jobs listed in the grid, and select particular jobs to be queued for batch processing. By default, AutoDock attempts to quickly find the best docking site by running 10 stochastic simulations per compound. The advantage of this is a fast run time completion, with the disadvantage being less accurate results when compared to using more simulations. Documentation suggests that AutoDock use at least 50 docking simulations to ensure results are accurate, however, it is also noted that results become increasingly better statistically as the number of dockings is increased. Literature has suggested that 100 simulations provides a good compromise between accuracy and speed [17]. This was the number chosen for DockoMatic’s default, although it can be increased or decreased in the text field labeled “AutoDock cycles” if more precise results, or quicker run times are the goal.
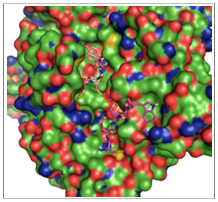
Below, in Figure 3.2, is an example of a DockoMatic result showing the best ranked (lowest binding energy) conformation for CCMWF in complex with Ac-AChBP as calculated by AutoDock.


Competing Binding Sites and Multiple Ligands
Typically, competitive binding experiments in AutoDock require a new protein complex receptor to be created from the results of an initial binding experiment. This complex receptor is generally comprised of the conformation with the lowest binding energy and the original receptor. This new receptor is then used with the other ligand in a conventional AutoDock job, resulting in an experiment showing how the second ligand may bind when the first is already bound to the same receptor. This type of experiment is time consuming and arduous with AutoDock, as it cannot automate the creation of the complex receptor nor start a second binding job automatically. Competitive binding experiments are completely automated in DockoMatic.
The GUIprovides a “Secondary Ligand” text box to enter the additional ligand used in the experiment. First the ligand from the standard “Ligand” text box is used in a normal AutoDock job. Then, DockoMatic creates a new protein complex from the original receptor and the highest ranking conformation from the first experiment. Next, the ligand from the “Secondary Ligand” text box is used to automatically create and run an AutoDock job with the new receptor. An example of two ligands docked to a receptor is shown in Figure 3.3; the results of a competitive binding experiment beginning with the docking of CCMWF to Ac AChBP receptor. The best result of CCMWF with Ac-AChBP forms a new receptor ligand complex. The secondary ligand, CDCMW, is then allowed to bind to the new complex. The lowest binding energy complex is displayed.
Ligand Creation
One requirement of AutoDock is that all ligand coordinate files to be submitted in .pdb format. The process of creating novel ligand structures can be tedious, and it is rarely the case that the .pdb files have been previously created. DockoMatic can be used to create peptide-based ligands as its sole duty, or as a prelude to an

AutoDock job [3]. I am not aware of any other software package that provides this functionality. DockoMatic automatically decodes a user supplied string of alphabet characters to produce a ligand .pdb file. Each character represents an amino acid, and when combined form a sequence for a ligand (e.g., GRWCK). Each of these characters maps to pre-built side chain .pdb files. The algorithm for creating a peptide ligand structure from the string representation can be summarized as follows.
1. add beginning
2. if next amino acid is not proline, add backbone structure
3. add amino acid sidechain
4. repeat steps 2 and 3 until the ligand string is exhausted
5. add end
6. optimize ligand structure
A hydrogen atom begins the chain of amino acids that form the ligand, while the chain ends with an oxygen and a hydrogen atom. The bend associated with the presence of proline in the backbone of a peptide or protein necessitates the one exception to the algorithm. Proline has the backbone already built into its side chain.pdb file, so when it is used, a backbone structure is not added to the chain. There are two static side chain .pdb files for each amino acid, providing representations in both an up and down orientation. Alternating sidechain-backbone pairs in the two orientations helps prevent atoms being set too closely together. An accounting of the static .pdb files reveals two .pdb files for each of the twenty amino acids, making 40, along with two backbone .pdb files for up and down, and the beginning and ending .pdb files comprise a library of 44 .pdb files used to create peptide-ligands.
Once the ligand structure is formed, a tool from the OpenBabel package, Obconformer, is used to optimize the structure [18]. Examples of pentapeptide ligands created with DockoMatic can be seen in Figure 3.4: A) CCMWF, B) CDCMW, C) CFWMW, D) CHMWW, and E) CHWWM.

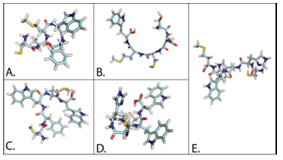
Cyclic Peptide Analog Creation
Cyclic peptide analog structures are mutations of ligands in which amino acids present in a structure scaffold are substituted. This process requires computations to be performed on the new 3D structure so that it is well-formed with respect to the orientation of the new amino acids to the backbone. Treepack was identified as software that already performs this task, so it was leveraged. DockoMatic accepts as input, the amino acids to be replaced and their replacements. In order for Treepack to effectively restructure the new amino acids, it must look at the relation of the two surrounding amino acids to the one being replaced and how differences in the structure of the new one will affect the rest of the sidechain. These three amino acids are excised from the ligand, and their identifiers are changed to reflect the new amino acid. Treepack then performs its computations on this tripeptide sidechain, and outputs the resulting sidechain. DockoMatic inserts the new sidechain back into the original peptide. This process is not limited to one substitution; DockoMatic uses a loop to perform these steps on as many substitutions as are specified.
Parallel Jobs on a Cluster
DockoMatic also facilitates running multiple jobs in parallel on a cluster by utilizing swarm [19,20]. Although DockoMatic was designed with the objective of being used on a cluster, it is not a requirement. Using an environment with multiple nodes allows jobs to be run independently and simultaneously, offering a linear speedup with regard to the number of nodes or cores.
Ranked Results
AutoDock outputs a single .dlg file containing the results of a docking job. Depending upon the number of simulation cycles used, this file can be quite large and unwieldy. Results are not easy to obtain, since the .dlg file either needs to be navigated manually, which is cumbersome, or viewed with AutoDock Tools. DockoMatic manages the .dlg file by parsing and summarizing results, providing a single file containing the binding energy, inhibition constant, conformation statistics, and cluster rank of each conformation listed in non-increasing order based on cluster rank. All resulting simulation conformations are also written to individual .pdb files, with the most favorable having the highest rank; 1 being the highest. The rank of 1 signifies the conformation with the lowest binding energy.
Result Analysis
Results can also be analyzed further. A second .gpf file can be supplied in conjunction with the “Analyze” option via a file browser window. This .gpf file would generally focus on a more limited area than the .gpf file used in the original docking experiment. The goal of this option is to evaluate how many conformations from a docking job fall inside and outside the new grid coordinates; useful when a binding domain is assumed based on experimental evidence. A text file is used to convey the results, and is comprised of the percent of runs that lie inside the secondary .gpf coordinates, average binding energy, best binding energy, average inhibition constant, and best inhibition constant. This feature can be of use in experiments where the goal is to find how likely it is for a ligand to bind to a particular site, when the entire receptor is available for the ligand to bind to. To conduct this type of analysis by hand would be laborious.
Molecular Modeling
TIM Wizard
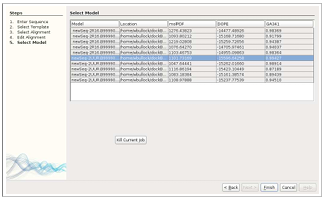
Model generation can be requested for ligands or receptors by checking the “Use Modeller” box. Next, when the appropriate button for ligand or receptor is pressed, the wizard TIM appears. TIM has five screens, each of which guide the user through a major step of the homology model creation process: Screen 1) enter sequence, output directory, and optional disulfide bond specifications (Figure 3.5), Screen 2) choose candidate templates for download (Figure 3.6), Screen 3) select alignments (Figure 3.7), Screen 4) modify alignments (Figures 3.8 and 3.9), and Screen 5) view resulting models (Figure 3.10).
Initial Input
TIM’s first window prompts the user to enter an output directory where the template and models are later stored. A large text area is also provided for the starting sequence to be entered manually. If a sequence .ali file containing the sequence already exists, this file location can be input instead, or browsed to by pressing the “Sequence” button.

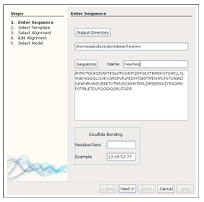
Template Selection
In TIM’s second step, the list of candidate templates is offered, in which the most desirable ones can be selected to be used for models. Candidate templates are found automatically using a BLAST search [21]. The list is populated with template name, e-value, length, score, identities, positives, and gaps. If yet more information is desired, a right-click of the mouse provides a pop-up window with the option to “Open in Browser” in which a Web browser is opened to the appropriate .pdb file on pdb.org. It is often the case that a given template includes multiple subunits, generally referred to as A, B, C, etc. If this is the case, DockoMatic lists the template as many times as there are subunits, specifying a different subunit with each entry.
This provides an easy way for the user to select which subunit they would like the alignment to be based on. In addition to using templates that were found with a BLAST search, the user can browse to existing templates using the “Browse for Template” button, in which the templates are added to the list.

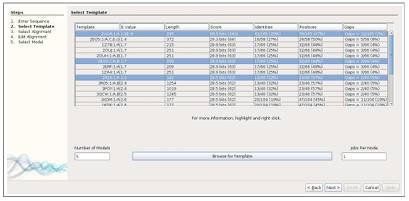
Sequence Alignment
Once the proper templates are selected, the sequence is aligned with the template using a modified version of MODELLER’s align2d.py script. The resulting alignments are displayed in the third window of TIM in list form with a one to one mapping to the selected templates.

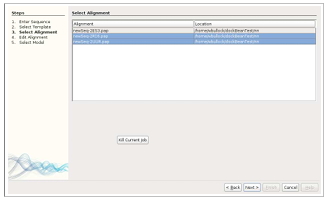
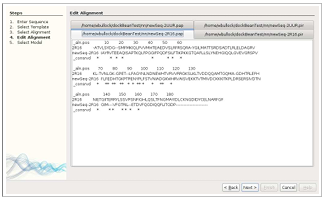
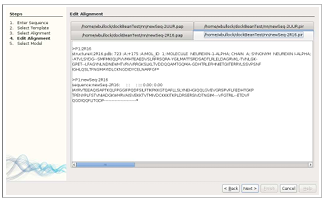
Alignment Modification
After the desired alignments are selected, TIM’s fourth window provides the user the option to manually modify the PIR and PAP alignment files in separate editor tabs. It is common to adjust an alignment to reflect desired structure elements before the modeling is performed. Generally, the two files are used together to determine what changes to make. The PAP file is used as a reference to easily find the locations for adjustments, while the PIR file is modified and used in the modeling experiment. If the alignment suffices as is, the user can simply press the “Next” button. The alignments are then used to generate 3D models.


Model Creation
In TIM’s fifth, and final window, models are generated and returned to the user in list form. The user selects which model is most appropriate for the type of experiment being conducted. The final number of models is dependent upon the number specified in “Number of Models” text field, the default being five. In addition to listing the models, the locations, MolPdf, DOPE, and GA341 values are presented to assist in determining and selecting the best model for the intended purpose. Once selected, the structure file path is automatically populated as a ligand or receptor to a binding experiment. When using disulfide bonds in the model, only the MolPdf score is available due to the information available from MODELLER output.

Disulfide Bond Creation
In DockoMatic, creating models with disulfide bonds does not require additional time. However, this task is not easy with MODELLER for beginning to novice users. If disulfide bonds are needed in the final structure being modeled, they can be specified in the “Residue Pairs” text box within the “Disulfide Bonding” area of TIM’s first window. The desired cysteine residue positions where the bond takes place is not limited to one set of positions; a colon separated list can be used to specify numerous disulfide bonds in the form of: “23-32:45-67:89-102.” TIM provides an example of such a list to avoid confusion.
Scoring and Evaluation of Alignments
For ease of selection of the final model, the MolPdf score is provided along with the DOPE and GA341 values, however, they are not a complete ranking system in and of themselves. The molpdf and DOPE scores cannot be used to rank models from different alignments, while the GA341 can. The GA341 score ranges from 0.0 to 1.0, where 0.0 is the worst, however, is not necessarily a good way to determine if a given model is better or worse than another. The user now has guidance on what model may be best for the intended use. If more information is needed, the locations of the models are provided for closer manual inspection of the .pdb files.
3.0.7 Performance
In order to utilize the full potential of DockoMatic, it should be used on a computing cluster, as it was designed. This allows one to manage large numbers of jobs at the same time, rather than being limited to local resources of a single computer. Using a cluster can give a linear increase in speed for the various types of experiments that can be performed. For instance, when creating homology models, typically a user must wait for MODELLER to run calculations for each model sequentially; if creating 100 models for a given sequence, in order to select the most favorable, MODELLER will calculate all 100 consecutively. DockoMatic creates all models simultaneously, giving significant benefits in speed; with 100 nodes, 100 models can be created on a cluster in the same time as one.
However, the benefits are not isolated to using a cluster. The creation of the homology model for the collagen α1 (XI) amino propeptide (Npp) protein with TIM in DockoMatic took a first time user close to six minutes, whereas the same experiment took fifty minutes when BLAST and MODELLER were run manually, along with the other steps necessary to create a homology model. While the two processes are similar, the main difference in times is due to the automation of an otherwise manual and time consuming series of steps. For instance, in the manual process, it took the user five minutes to visit the NCBI database, enter the sequence for the target protein, select the correct options, run the BLAST search, and to select and download the zip file for the correct template structure. This contrasts with DockoMatic’s one minute, where all that is required is to enter the sequence in TIM’s text box, hit the button, and when the results return, select the desired template. The rest of the 45 minutes for the manual method was used to unzip the template file, creating a “.ali” file for the target protein, editing the MODELLER script, called “align2d.py” to accommodate the new settings, creating file labels and saving project files in accordance with MODELLER tutorial instructions, and initiating model creation. These steps were all automated with DockoMatic, and took an additional four minutes.
3.0.8 Publications
I currently have two publications for DockoMatic. The first, “DockoMatic- au tomated ligand creation and docking,” was published in BMC Research Notes in 2010 [3]. This paper introduces DockoMatic, outlining the features that reinforce it as a powerful tool capable of significantly reducing the complexity of managing multiple AutoDock jobs. The second, “DockoMatic: Automated peptide analog creation for high-throughput virtual screening,” was published by the Journal of Computational Chemistry in 2011 [22]. This publication documents the addition of cyclic peptide analog creation and evaluates the high-throughput capabilities of DockoMatic.
Another manuscript is currently under review by the American Chemical Society Journal of Chemical Informatics and Modeling. It outlines new features for DockoMatic, including homology modeling with TIM, Inverse Virtual Screening, exchangeable docking engines, and an all new GUI designed with NetBeans.
3.0.9 Research Projects
DockoMatic has proven to be useful and is actively being used in numerous ways on and off the Boise State University campus. One appealing aspect of DockoMatic is that it cuts down the time for novice chemistry and biochemistry students to get to the point of performing experiments, since they no longer have to learn involved
scripting and the idiosyncrasies of existing software suites. Presentations in the form of live tutorials and workshops have been used to get these students familiar with DockoMatic, molecular docking, and modeling.
One tutorial outlines the creation of homology models, and uses the collagen (XI) α1 amino propeptide protein as an example. This type of experiment took a novice undergraduate chemistry student three months to gain the experience to create the model without DockoMatic. With DockoMatic, users are able to perform this experiment in under 10 minutes. Another tutorial taught students to use DockoMatic for high-throughput molecular mutation and binding experiments. The goal of this experiment is to analyze α-conotoxin TxIA bound to the acetylcholine binding protein from Aplysia californica. First students analyze the protein complex and propose favorable interactions. Next, the ligand is removed, the receptor is cleaned, and a ligand mutation is recommended. The resulting ligand is bound, and students identify whether the result is consistent with the prediction. This is an important step for students wishing to gain familiarity in the realm of molecular docking, and DockoMatic makes their journey easier and less time consuming.
As well as being currently used, it has also aided with projects in the recent past. Members of the Colorado School of Mines, under the guidance of Dr. C. Mark Maupin, have used DockoMatic to perform the virtual screening of acetylcholine and α-conotoxin MII to the α3β2 nicotinic acetylcholine receptor homology model. DockoMatic has also been used in evaluational studies of high-throughput virtual screening tools [23].
CHAPTER 4
CONCLUSIONS
I have developed novel algorithms in the field of Bioinformatics, with publications supporting such work. The tools I have created offer a linear speedup over more traditional techniques. CseqStat speeds up processing the large amounts of sequencing data for nu cleotides and proteins in the NCBI database. This application is robust in its abilities, allowing one to find the frequency of all sequences of a given length, store the results in an efficient manner, and provide a mechanism for quick and easy retrieval of such data, either from the command-line, or from a client-server GUI.
DockoMatic has been developed as a solitary tool to aid in numerous aspects of Bioinformatic study. It provides a user-friendly GUI to ease the management of molecular docking jobs, as well as facilitate the automatic creation of peptide ligands. With the wizard TIM, homology model creation is vastly simplified and sped up over conventional methods. It also provides an easily extendable and customizable interface, as it has been developed with the NetBeans framework, offering the ability to undock and resize individual window components of the GUI for an infinitely configurable work environment. Fledgling chemists, as well as experts, can benefit from DockoMatic’s simple interface and powerful speed.
REFERENCES
[1] Alileche A, Goswami J, Bourland W, Davis M, Hampikian G: Nullomer derived anticancer peptides (NulloPs): Differential lethal effects on normal and cancer cells in vitro. Peptides 2012, [http://www.sciencedirect. com/science/article/pii/S0196978112%004044].
[2] Kitchen D, Decornez H, Furr J, Bajorath J: Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery 2004, 3:935–949.
[3] Bullock C, Jacob R, McDougal O, Andersen T, Hampikian G: DockoMatic automated ligand creation and docking. BMC Research Notes 2010, 3:289.
[4] NCBI [http://www.ncbi.nlm.nih.gov/].
[5] Hindman L: Designing Reliable High-Performance Storage Systems For HPC Environments. Master’s thesis, Boise State University, Computer Science Department 2011.
[6] Jacob R, McDougal O: The M-superfamily of conotoxins: a review. Cellular and molecular life sciences 2010, 1:17.
[7] Millard E, Daly N, Craik D: MINIREVIEW: Structure-activity relation ships of alpha-conotoxins targeting neuronal nicotinic acetylcholine receptors. European Journal of Biochemistry 2004, 271(12):2320–2326.
[8] Zoete V, Grosdidier A, Michielin O: Docking, virtual high throughput screening and in silico fragment-based drug design. Journal of Cellular and Molecular Medicine 2009, 13(2):238–248.
[9] Kontoyianni M, McClellan LM, Sokol GS: Evaluation of Docking Performance: Comparative Data on Docking Algorithms. Journal of Medicinal Chemistry 2004, 47(3):558–565, [http://pubs.acs.org/doi/abs/10.1021/ jm0302997]. [PMID: 14736237].
[10] Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Ol son AJ: Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry 1998, 19(14):1639–1662, [http://dx.doi.org/10.1002/ (SICI)1096-987X(19981115)19:14<163%9::AID-JCC10>3.0.CO;2-B].
[11] Cavasotto C: Ligand docking and virtual screening in structure-based drug discovery. In From Physics to Biology. Edited by Clemente-Gallardo J, Moreno Y, lorenzo JS, Velasquez-Campoy A 2006:34–49.
[12] TreePack [http://ttic.uchicago.edu/~jinbo/TreePack.htm].
[13] Modeller [www.modeller.com].
[14] PyMOL [http://www.pymol.org].
[15] NetBeans IDE [http://netbeans.org/].
[16] AutoDock Tools [http://autodock.scripps.edu/resources/adt/index_ html].
[17] Li C, Xu L, Wolan DW, Wilson IA, Olson AJ: Virtual Screening of Human 5-Aminoimidazole-4-carboxamide Ribonucleotide Trans formylase against the NCI Diversity Set by Use of AutoDock to Identify Novel Nonfolate Inhibitors. Journal of Medicinal Chemistry 2004, 47(27):6681–6690, [http://pubs.acs.org/doi/abs/10.1021/ jm049504o]. [PMID: 15615517].
[18] OpenBabel [http://openbabel.sourceforge.net/].
[19] Swarm-Java [http://cs.boisestate.edu/~amit/research/swarm/].
[20] Swarm [http://biowulf.nih.gov/apps/swarm.html].
[21] BLAST [http://blast.ncbi.nlm.nih.gov/Blast.cgi].
[22] Jacob R, Bullock C, Andersen T, McDougal O: DockoMatic: Automated peptide analog creation for high throughput virtual screening. Journal of Computational Chemistry 2011, 32:2936–2941.
[23] Jacob RB, Andersen T, McDougal OM: Accessible High-Throughput Virtual Screening Molecular Docking Software for Students and Educators. PLoS Comput Biol 2012, 8(5):e1002499, [http://dx.doi.org/10.1371% 2Fjournal.pcbi.1002499].